Workflows
MassChemSite provides 3 different workflows for the different chemical applications.
Derivatization
Derivatization workflow is the simplest one. Accepts an unique compound as reactant and is aimed for derivatization reactions. The areas of interest of this workflow are organic synthesis procedures like late stage functionalization,(10) animal or plant based metabolism,(12-14) force degradation studies among others. To illustrate the usability of this workflow, an example of Late Stage Functionalization application is described below.
Late-stage functionalization (LSF) is a chemical emerging as a trans-formative approach for drug discovery.(11) In LSF, the C-H bonds of drug leads or advanced intermediates are subjected to a number of chemical reactions in order to generate new analogues in few synthetic steps and therefore avoiding the time-consuming de Novo syntheses. In the way a great number of compounds could be obtained in order to rapidly understand structure-activity relationships (SAR), target selectivity, potential off-target liabilities, the metabolic soft spots and/or produce chemical biology probe molecules.
However, the strong point of accessing a great number of potential compounds from the derivatization of C-H bonds can also be one of the technique weak points since each reaction can lead to the formation of multiple regioisomeric products. The separation, purification and characterization of these closely related regioisomers is often slow and laborious, and therefore making LSF approaches less attractive that one could imagine. Therefore it is necessary to have in hand an efficient, simplified and if possible automatic analytical work flow that can provide structural information on the regiochemistry of C–H reaction starting from a crude reaction mixture prior to isolation. In this way the more laborious process of purification and structure confirmation could be done only in those samples with the highest-value.
The problem of identifying (finding and assigning the chemical structure) regioisormers from a chemical reaction is not very different to the one found in drug discovery for the metabolite identification. High-resolution MS/MS techniques have long been employed in pharmaceutical research to determine sites of oxidative drug metabolism mediated by cytochrome P450 oxygenase enzymes.(1) These reactions typically involve the abstraction of an hydrogen atom and its replacement by an hydroxyl group, which is very similar to a C-H Functionalization done in LSF.
The comparison of the MS/MS fragmentation pattern of the parent molecule and the metabolites yield the site of metabolism (SoM). This process that typically was done manually and it was time consuming has recently being highly automatized using sophisticated data analysis software algorithms that aims to do the interpretation of tandem mass spectrometric data and assignment of individual fragment ions based on accurate mass and atom connectivity.(1-9) Due to the obvious synthetic similarities between metalloenzymecatalyzed C–H oxidation reactions and modern C–H functionalization methodologies, similar software algorithms, adapted to accommodate a broader array of C–H bond transformations, could be used to help in the structural elucidation for LSF. Finally these mass spectra derived techniques typically cannot get a single potential structure, since fragmentation of the molecules may not allow the discrimination between close atoms in the same molecular region. In this case the NMR prediction techniques could be used in order to select which of the proposed structures is the one compatible with the experimental spectra. One of the advantages of the analytical technique used is that it can work with very low concentrations, in general the metabolite concentrations that nowadays could be detected and identified are in the order of nano molar. Nevertheless in LSF the aim is to obtain a major product or a small list of potential products therefore, the peak finding is simpler in the LSF case than in metabolite identification.
An example is shown for the chlorination of Risperidal in order to illustrate the strategy that can be followed in MassChemSite for LSF. Risperidal is a complex molecule that contains 27 C-H positions where a chlorination, replacement of a C-H bond by a C-Cl bond, could happen. The chlorinated compounds could be classified in 4 groups depending on the region of the chlorination as shown in the next figure.
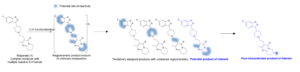
Background-Late-Stage Functionalization: Risperidal example. Adapted from Yao H, et al
Background-Late-Stage Functionalization: Risperidal example. Adapted from Yao H, et al
The chlorination reaction can be done under multiple conditions with different reactants and solvents. If one would like to isolate all the potential product to understand the best conditions to obtain the different regioisormers it would be a laborious task. Instead, this task could be simplified by using a synthesis in a plate form:
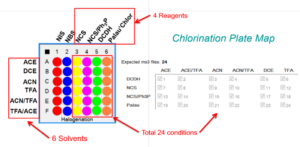
96 well plate experiment. Four compounds per plate
The experiments are done, and the plates are submitted directly to LC-MS analysis and MassChemSite process, yielding the following summary of products:
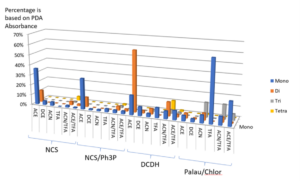
Results from the chlorination. Adapted from K. Bateman presentation at the CPSA USA 2016
In addition to the quantitative information, also a structural information is obtained for each reaction product:
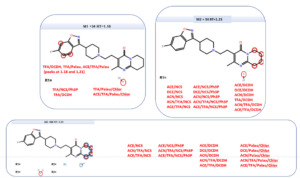
Structural elucidation from MassChemSite. Analysis of the multiple conditions.
Adapted from K. Bateman presentation at the CPSA USA 2016
Therefore MassChemSite automatically finds a number of products related to the initial compound that undergoes one or more than one chlorination and it is able to localize the positions/regions where the reaction actually happens and also the experimental conditions that better produce the different compound of interest.
Chemical Monitoring
In the Chemical Monitoring (CM) workout the user can monitor a reaction that has multiple reactants and multiple products. In this strategy MassChemSite is looking for the compounds that can be declared in a reaction file (Rxn) or a structure file (sdf), but also the derivatives that can be produced for any of the compounds using the reactions that are selected by the user. In this way the system could look for expected by-products. These are compounds which formation could be foreseen from the chemicals in the reaction media and the set of reactions made by the user. In order to show this innovative workflow, the reaction of the coupling of the 4,6-dichloro-5-Methylpyrimidine with the Phenylboronic acid in toluene/methanol and using as capitalizer the Palladium-triphenylphosphine (1:4) salt will be shown.
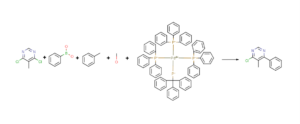
Chemical Monitoring: Example of the chemical monitoring input file
The user introduces the reaction into the system declaring all the reactants and expected products, but also the workflow enables the possibility to add custom reactions that can produce potential chemical modifications in the reaction mixture, like the ones shown in the following figure:
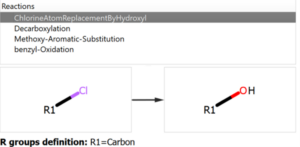
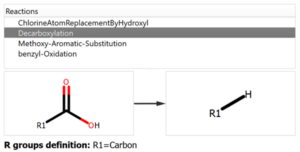
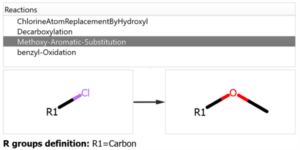
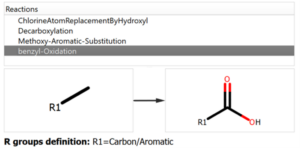
Chemical Monitoring example: Custom Reaction definition
After the reactants/products, other potential reactions and the raw data file for the LC-MS acquisition of the reaction crude after 19 hours of reaction are introduced into MassChemSite and the software has processed the data the following result is obtained:
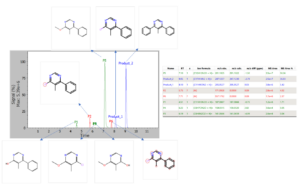
Chemical Monitoring: Example for the data analysis for the LC-MS data obtained from the reaction crude after 19 hours of reaction.
In this analysis 8 different chromatographic are obtained that correspond to 8 products of the reactions, since no reactant was left after the 19 hours.
Product_1 and Product_2: correspond to the desired reaction between the Chlorine atom and the boronic acid. The Product_2 is the double reaction and probably indicates that the reaction mixtures was let to react too long time.
P1: It is a product from the reaction mixture that is not one of the expected products (Product 1 or product_2) but it could be obtained by the replacement of the Chlorine atom by an hydroxyl group. The chemical pathway to get this product is no known, since it could come from P5 and hydrolysis of the ether formed or it could be an aromatic substitution of the chlorine by an hydroxyl group. This product could be identified because the chemical reaction was added into the pool of reactions to be used (see previous figures).
P5: It is another major non expected product of the reaction that could come from the replacement of the chlorine atom but a methoxy group that may come from methanol used as solvent. This by-product is the major one obtained in the chemical conditions applied. Again, since there is no analysis of the time course of the reaction, it is not possible to know what is the mechanism, if the reaction first form the methoxy derivative in one of the two chlorine atoms of the starting material and the reaction of the other chlorine atom with the boronic acid or the opposite. In case of having kinetic data, the evolution of the P5 and product_1 could give us a hint on the chemical pathway to get such a by-product. This product could be identified because the chemical reaction was added into the pool of reactions to be used (see previous figures).
P3 and P4: These two unexpected products are obtained as initial reaction of one of the chlorine with the methanol in the media and ulterior hydrolysis. These products could be identified because the chemical reaction was added into the pool of reactions to be used (see previous figures).
P2: this compound appears as a red peak which means that the software was able to detect the presence of the compound but it could not identify its structure. Nevertheless, the software perform the fragmentation analysis to relate the compound to any of the reactants and/or expected products and it was able to identify that the chemical modification occurs in the Chlorine. An analysis of the difference in the m/z values for the parent and the detected peak reveals that probably it is an aromatic reduction of the chlorine atom, replacing the Chlorine atom by an hydrogen one. The structure of this product could not be assigned since that chemical reaction was not introduced in the analysis, although the software correctly interpret the spectra an localize the chemical modification on the Chlorine atom.
P6: This detected peak is also an unknown structure (red peak), but in this case the system can not localize the place in the reactants that may lead to the m/z shift observed for this compound and therefore it marks all the atoms in the molecule as potential places for chemical modifications.
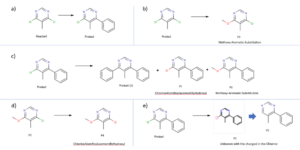
Chemical Monitoring: summary of the results found in the analysis of the reaction crude after 19 hour.
a) correspond to the expected compounds, b) Reaction of the initial reactant to yield a methoxy derivative, c) reactions that undergo the expected compound, d) further derivatization of the methoxy derivative obtained in reaction b, e) modification of the wanted product to an unknown with the structural modification on the chlorine atom.
Reaction Tracking
Reaction Tracking (RT) workflow add an extra level to the reaction centered workflows adding the possibility to perform multi-member reactions enhancing the identification of by-products.

Reaction tracking allows the user to add reactions involving more than one reactant.
This feature allows to the user customize reactions to combine both given reactants and virtually created ones in order to find all possible products given by the combination of all chemical entities. Additionally, it provides to MassChemSite the possibility to generate intramolecular reactions and reactions with solvents.
To illustrate the use of Reaction Tracking workflow, the application of MassChemSite to identify by-products in synthesis of PROTACs will be described below.
PROTACs are heterobifunctional small molecules composed of a ligand for a protein of interest (POI) and an E3 ligase recruiter connected through a linker. Instead of inhibiting the protein functions, PROTACs promote the formation of a ternary complex with POI and E3 ligase, inducing POI poly-ubiquitylation and its successive proteasomal-dependent degradation. This appealing technology has already attracted great attention from both academia and industry, and the optimization of PROTACs’ synthetic procedures is now needed. (15)
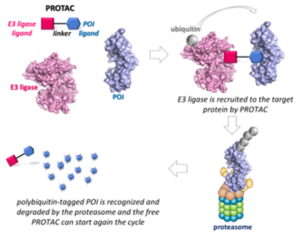
General description of PROTACs. Paiva, S.-L.; Crews, C.M. Curr. Opin. Chem. Biol. 2019, 50, 111-119.
The reaction shown in the scheme below represents the final step for the synthesis of PROTAC under study. In particular, the POI ligand functionalized with linker (intermediate 1) (1.0 equiv) and fluorothalidomide (2) (1.0 equiv) were reacted in presence of N,N-diisopropylethylamine (DIPEA) (3.0 equiv) in DMF at 90° C for 24h. During the reaction time, samples at time 15 and 60 minutes were collected and analyzed by LC-HRMS.
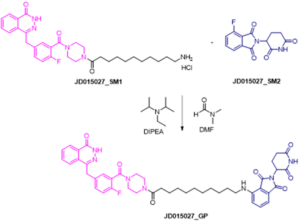
Final step of the synthesis of PROTAC under study. JD0150127_SM1, JD015027_SM2, DIPEA and DMF were used as input compounds.
After processing the raw file in MassChemSite using the reaction tracking workflow, the expected product (m/z = 806,3672) was found at RT 5.5min in both samples and its structure was assessed by MS2:
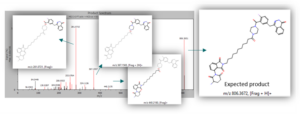
MS2 of the expected product and structure assignation of key fragments
In addition to the product of interest, MassChemSite also detected some byproducts, identifying them in most of the cases (blue: reactant, green: byproduct, red: unknown)
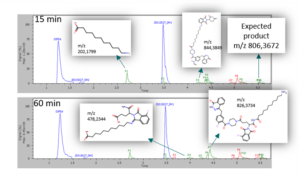
Reaction by products found at time 15 and 60 min
The origin of those byproducts is the reaction of hydrolysis products (m/z 202,1799) with one of the reactants (m/z 478,2344) of both (m/z 844.3849) of the reactants. Also an unexpected product due to a prior ring opening of one of the products has been found (m/z 826,3734).
- Enhanced metabolite identification with MS(E) and a semi-automated software for structural elucidation. Bonn B, Leandersson C, Fontaine F, Zamora I. Rapid Commun Mass Spectrom. 2010 Nov 15;24(21):3127-38
- Update on hydrocodone metabolites in rats and dogs aided with a semi-automatic software for metabolite identification Mass-MetaSite. Li AC, Chovan JP, Yu E, Zamora I. Xenobiotica. 2013 Apr;43(4):390-8.
- Software automation tools for increased throughput metabolic soft-spot identification in early drug discovery. Zelesky V, Schneider R, Janiszewski J, Zamora I, Ferguson J, Troutman M. Bioanalysis. 2013 May;5(10):1165-79.
- High-throughput, computer assisted, specific MetID. A revolution for drug discovery. Zamora I, Fontaine F, Serra B, Plasencia G. Drug Discov Today Technol. 2013 Spring;10(1):e199-205
- Software-aided structural elucidation in drug discovery. Ahlqvist M, Leandersson C, Hayes MA, Zamora I, Thompson RA. Rapid Commun Mass Spectrom. 2015 Nov 15;29(21):2083-9.
- Post-acquisition analysis of untargeted accurate mass quadrupole time-of-flight MS(E) data for multiple collision-induced neutral losses and fragment ions of glutathione conjugates. Brink A, Fontaine F, Marschmann M, Steinhuber B, Cece EN, Zamora I, Pähler A. Rapid Commun Mass Spectrom. 2014 Dec 30;28(24):2695-703
- Software-aided cytochrome P450 reaction phenotyping and kinetic analysis in early drug discovery. Cece-Esencan EN, Fontaine F, Plasencia G, Teppner M, Brink A, Pähler A, Zamora I. Rapid Commun Mass Spectrom. 2016 Jan 30;30(2):301-10.
- Development, optimization and implementation of a centralized metabolic soft spot assay. Paiva AA, Klakouski C, Li S, Johnson BM, Shu YZ, Josephs J, Zvyaga T, Zamora I, Shou WZ. Bioanalysis. 2017 Apr;9(7):541-552.
- Software-aided approach to investigate peptide structure and metabolic susceptibility of amide bonds in peptide drugs based on high resolution mass spectrometry. Radchenko T, Brink A, Siegrist Y, Kochansky C, Bateman A, Fontaine F, Morettoni L, Zamora I. PLoS One. 2017 Nov 1;12(11):e0186461
- Enabling Efficient Late Stage Functionalization of Drug-Like Molecules with LC-MS and Reaction Driven Data Processing. Yao H, Liu Y, Tyagarajan S, Streckfuss E, Reibarkh M, Chen K, Zamora I, Fontaine F, Goracci L, Helmy R, Bateman KP, Krska SW, Eur JOC, DOI: 10.1002/ejoc.201701573
- The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Cernak T, Dykstra KD , Tyagarajan S, Vachal P, Krska SW, Chem. Soc. Rev. 2016, 45, 546.
- Degradation studies of dimethachlor in soils and water by UHPLC-HRMS: putative elucidation of unknown metabolites. López-Ruiz R; Romero-González R; Ortega-Carrasco E; Martínez Vidal JL; Garrido Frenich A, Pest. Manag. Sci. 2020, 76(2), 721-729.
- Dissipation kinetic studies of fenamidone and propamocarb in vegetables under greenhouse conditions using liquid and gas chromatography coupled to high-resolution mass spectrometry López-Ruiz R; Romero-González R; Serra B; Garrido Frenich A, Chemosphere 2019, 226, 36-46.
- Dissipation studies of famoxadone in vegetables under greenhouse conditions using liquid chromatography coupled to high-resolution mass spectrometry: putative elucidation of a new metabolite. López-Ruiz R; Romero-González R; Ortega-Carrasco E; Garrido Frenich A, J. Sci. Food Agri. 2019, 99(12), 5368-5376
- Understanding the Metabolism of Proteolysis Targeting Chimeras (PROTACs): The Next Step toward Pharmaceutical Application. Goracci L; Desantis J; Valeri A; Castellani B; Eleuteri M; Cruciani G, Journal of Medicinal Chemistry, 2020, 63(20), 11615–11638.