

74th ASMS Annual Conference 2026 – POSTERS
Mass Analytica posters at 74th ASMS Annual Conference 2026

74th ASMS Annual Conference 2026
Come visit us at Booth #617!
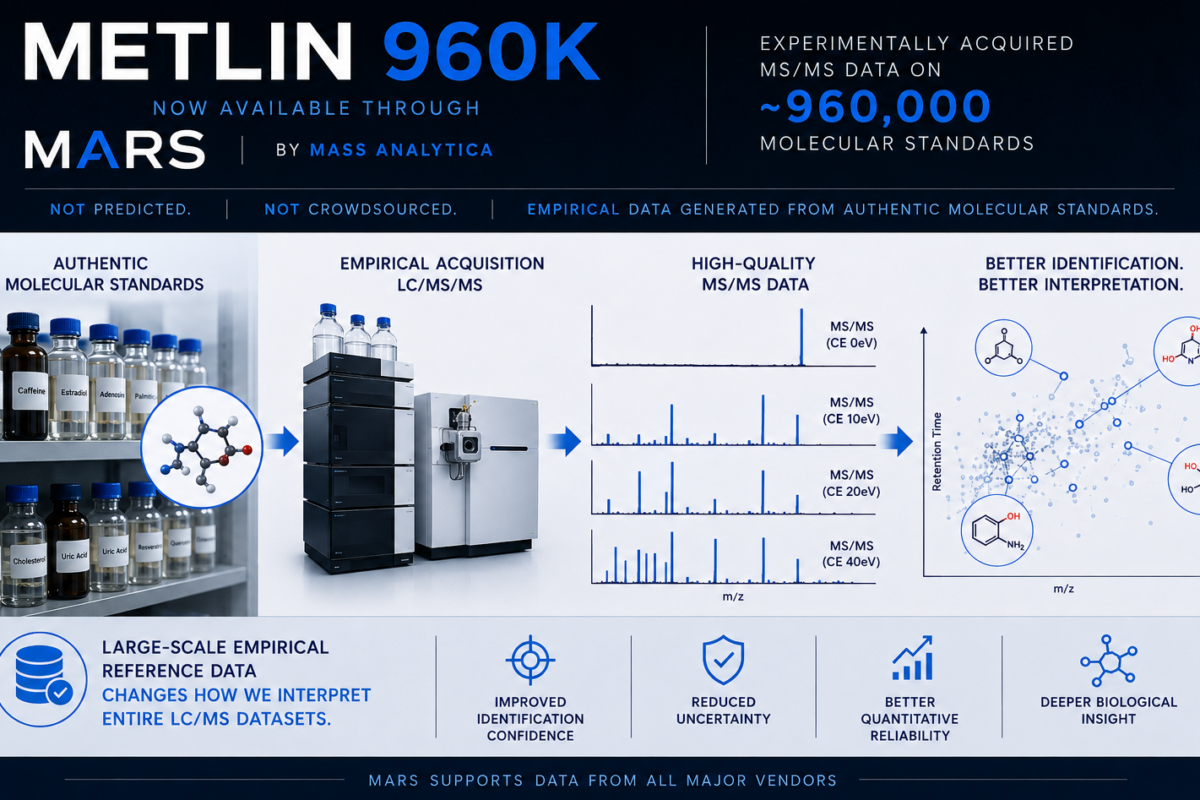
MARS 1.1.0 release note
We are glad to announce that we recently released MARS 1.1.0!
SEAMLESS SUPPORT FOR OLD AND NEW LIPID NOMENCLATURE IN LIPOSTAR2
You must be logged in to access this content. Not yet registered? Create a new account